

Abstract
We have developed small peptide mimetics of IFN-γ that can bypass the poxvirus virulence factor B8R protein, which binds to intact IFN-γ and prevents its interaction with receptor extracellular domain. Thus, these peptides inhibit vaccinia virus replication in cell culture where intact IFN-γ is ineffective. We demonstrate here that the mouse IFN-γ-mimetic peptide, IFN-γ95–132, protects C57BL/6 mice against overwhelming lethal vaccinia virus infection. The mimetic peptide was synthesized with an attached lipophilic group for penetration of cell plasma membrane. Injection of mimetic i.p. before and at the time of intranasal (106 PFU) or i.p. (107 PFU) challenge with virus resulted in complete protection at 200 μg of mimetic and 40–60% protection at 5 μg of mimetic. Initiation of treatment of mice with IFN-γ mimetic up to 2 days postinfection resulted in complete protection against death, whereas initiation of treatment at 6 days postinfection resulted in 40% protection. Administration of mimetic by the oral route also completely protected mice against the intranasal route of a lethal dose of vaccinia virus challenge. In addition to its direct antiviral effect, the mimetic also possessed adjuvant effects in boosting humoral and cellular immunity to vaccinia virus. The combination of antiviral and adjuvant effects by the IFN mimetic probably plays a role in its potent anti-vaccinia virus properties. These results suggest an effective therapeutic against ongoing, lethal poxvirus infections that taps into innate and adaptive host defenses.
The IFNs are the primary players in host innate and adaptive immune responses against viruses (1). There are two main classes of IFNs, type I and type II (2). Type I IFNs are classified into various related types, IFN-α, with at least 14 contiguous genes, a single β, several IFN-τ, IFN-ω, and other recently discovered types (3). Type II IFN, by contrast, has only one member, called IFN-γ (2). Despite the various type I IFNs, they all recognize the same hetero-oligomeric receptor consisting of IFNAR1 and IFNAR2 chains, whereas IFN-γ binds to a different receptor consisting of the chains IFNGR1 and IFNGR2 (4).
The general view is that IFNs, both type I and type II, exert their effects by cross-linking the receptors and activation of the JAK/STAT transcription pathways, STAT1α/STAT2 heterodimer for type I IFNs and STAT1α/STAT1α homodimer for type II IFN (5, 6). This in turn results in activation of at least three key antiviral pathways: protein kinase R, 2′-5′ oligoadenylate synthetase, and RNA-specific adenosine deaminase (2). The net effect of these activations is to interfere with synthesis of virus proteins.
Viruses have developed a variety of mechanisms to inhibit or blunt the antiviral apparatus of the IFN system. Poxviruses are particularly astute at thwarting the IFN system. These are large double-stranded viruses that replicate in the cytoplasm of the cell. The variola strain of the poxviruses is responsible for smallpox, which historically has been the causative agent of pandemics that have resulted in considerable loss of human life (7). Worldwide, it is estimated that smallpox has killed up to 500 million people in the 20th century, and ∼400,000 annually in Europe in the 18th century. Modern medicine has responded to smallpox by global immunization, but this has been discontinued for many years, which has basically resulted in lack of protection of the entire global community to natural or deliberate reintroduction of the virus into society. In addition, the emergence of new poxviruses by recombination within this family of viruses, or by their crossing the species barrier, such as the one seen recently in the outbreak of monkeypox virus in Africa and the mid-Western United States (8), are strong reasons for developing effective therapies against this family of viruses.
The assembly of poxviruses in the cytoplasm of infected cells is complex, involving the generation of four types of infectious virus particles (7). Attachment, internalization, and disassembling of poxviruses is followed by initiation of three waves of mRNA synthesis. The first or early wave codes for virus growth factors and decoy cytokine receptors. Decoy receptors for both type I and type II IFNs are produced during early protein synthesis in poxvirus-infected cells. An important virulence factor of poxviruses is the B8R protein, which is a homolog of the extracellular domain of the IFN-γ receptor and can therefore bind to intact IFN-γ and prevent its interaction with the receptor (9).
It is desirable to develop IFN-γ-like drugs that have direct antiviral activity as well as innate and adaptive immune function in the presence of virulence factors such as the B8R protein of poxviruses such as vaccinia. Related to this objective, we have discovered, characterized and synthesized small peptide agonists/mimetics of IFN-γ (10, 11, 12). These peptides, consisting of murine IFN-γ (mIFN-γ)395–132) for mice and human IFN-γ95–134 for humans, possess potent antiviral activity in tissue culture against vaccinia virus, encephalomyocarditis virus, and vesicular stomatitis virus (12). The peptide mimetics act intracellularly to activate the JAK/STAT signaling apparatus and do not recognize the IFN-γ receptor extracellular domain (12). Thus, vaccinia virus virulence factor B8R did not interact with the IFN-γ mimetics and failed to block mimetic inhibition of vaccinia virus and B8R-expressing encephalomyocarditis virus replication in culture (12). The mimetics were delivered into the cell via penetration by attachment of a lipophilic group, palmitate, to chemically synthesized peptides as described by us (11, 12). This report determines the ability of the IFN mimetics to protect mice against overwhelmingly lethal doses of vaccinia virus, even at late stages of infection.
Materials and Methods
Cells and virus
BSC-40 cells were obtained from American Type Culture Collection and propagated on DMEM with 10% FBS. All cells were grown at 37°C in humidified atmosphere with 5% CO2. Vaccinia virus Western Reserve was a gift from Dr. Richard Condit (University of Florida, Gainesville, FL). Vaccinia virus was grown, titrated on BSC-40 cells, and purified on sucrose gradient, as described (13).
Peptides
Peptides corresponding to residues 95 through 132 of murine IFN-γ, IFN-γ95–132(Lipo-AKFEVNNPQVQRQAFNELIRVVHQLLPESSLRKRKRSR), and the one corresponding to residues 95 through 125, IFN-γ95–125 (Lipo-AKFEVNNPQVQRQAFNELIRVVHQLLPESSL) were synthesized on an Applied Biosystems 9050 automated peptide synthesizer using conventional fluorenylmethyloxycarbonyl chemistry as described previously (10). The addition of a lipophilic group (palmitoyllysine) to the N terminus of the synthetic peptide was performed as a last step, using semiautomated protocol. Peptides were characterized by mass spectrometry and were purified by HPLC.
Mice
All animal protocols were approved by the Institutional Animal Use and Care Committee at the University of Florida. Female C57BL/6 mice (6–8 wk old) were purchased from The Jackson Laboratory. Peptides dissolved in PBS in a volume of 100 μl were administered i.p. For the oral administration of the peptides, indicated amounts of peptide in 0.5 ml of PBS were given using a feeding needle. Vaccinia was administered i.p. in a volume of 100 μl. For intranasal administration, vaccinia virus was taken in a volume of 10 μl, and 5 μl were delivered in each of the nostrils of a lightly anesthetized mouse. After infection, mice were observed daily for signs of disease, such as lethargy, ruffled hair, weight loss, and eye secretions. Moribund mice were euthanized and counted as dead.
Measurement of anti-vaccinia Ab response by ELISA
Microtiter plates were coated with 106 PFU of purified UV-inactivated vaccinia virus (900,000 μJ/cm2 for 5 min in a DNA cross-linker) in 100 μl of binding buffer (carbonate-bicarbonate, pH 9.6) overnight at 4°C. Plates were blocked for 2 h at room temperature with PBS containing 5% FBS. Mouse sera (n = 5) were serially diluted in PBS containing 0.1% Tween 20 (wash buffer); 0.1 ml of the diluted serum was added to each well. The plate was incubated for 2 h at room temperature and washed three times with wash buffer. Peroxidase-conjugated goat anti-mouse IgM (μ-chain specific), or IgG (γ-chain specific), both from Santa Cruz Biotechnology, diluted at 1/2000 in a volume of 0.1 ml was added to each well, incubated for 1 h, and washed five times with wash buffer. o-Phenylenediamine (Pierce) in a volume of 0.1 ml was added and incubated for 15 min. The reaction was stopped by addition of 50 μl of 3 N HCl. The OD490 was determined using a microtiter plate reader.
Measurement of vaccinia virus-neutralizing Abs
Plaque reduction assay was conducted to test the ability of Abs to inhibit viral infection of target cells. BSC-40 cells were seeded to confluency in a six-well plate the day before the assay. Sera obtained from mice (n = 5) on days indicated were heated at 56°C for 30 min to inactivate the complement. Purified vaccinia virus (100 PFU) was incubated with a known dilution of serum at 37°C for 1 h, followed by addition to BSC-40 cells. After 1 h, the virus-containing medium was replaced with fresh medium containing 0.5% agarose and 0.01% neutral red. Three days later, the number of plaques was counted. The number of plaques in wells with vaccinia alone was taken as 100%. Percent reduction in other treatments conducted in triplicate was measured and is presented as average with SD.
Measurement of vaccinia-specific cellular response by proliferation assay
Spleens from naive or recovered mice at times indicated were homogenized to single-cell suspension. Splenocytes (105 cells/well) were incubated with medium alone or with medium containing UV-inactivated vaccinia virus at 37°C for 96 h. The cultures were then pulsed with [3H]thymidine (1 μCi/well; Amersham Biosciences) for 8 h before harvesting onto filter paper discs using a cell harvester. Cell-associated radioactivity was counted using a scintillation counter. In the experiment described in Fig. 5 a, Alamar blue dye assay (14) was used to test the proliferation of splenocytes.
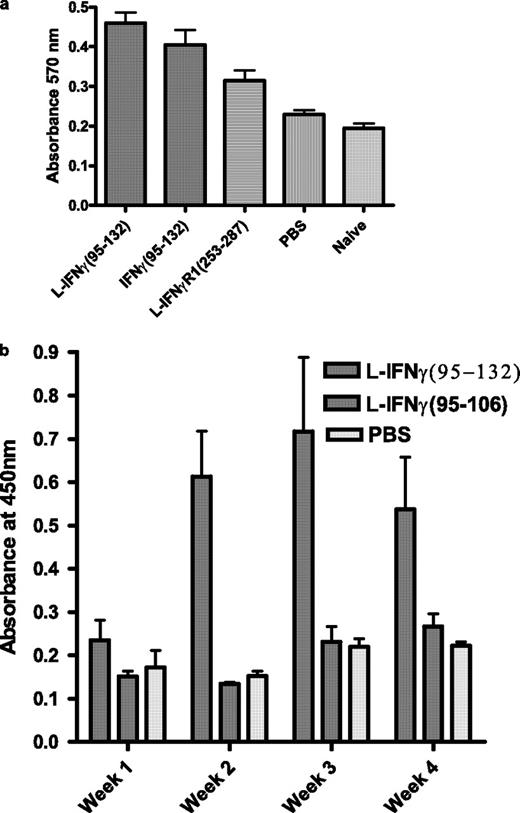
Adjuvant effect of IFN-γ mimetic for vaccinia virus and BSA response. a, Mice (n = 5) were infected intranasally with vaccinia virus in the presence of lipo (L)-IFN-γ95–132, IFN-γ95–132, control peptide, or PBS. Proliferation in the presence of purified inactivated vaccinia virus was determined 48 h later by using Alamar blue dye (25 ). Statistical measurements using the Wilcoxon-Mann-Whitney rank sum test indicated p < 0.05 for mimetic vs control. b, Mice (C57BL/6, n = 5) were immunized using BSA as an Ag in the presence of lipo-IFN-γ95–132, control peptide, or PBS. On the weeks indicated, blood was drawn from mice and measured for the presence of BSA-specific Abs in an ELISA format. The values represent the average with SD. p < 0.05 was obtained for the lipo-mimetic- vs the PBS-treated group.
Adjuvant effect of IFN-γ mimetic for vaccinia virus and BSA response. a, Mice (n = 5) were infected intranasally with vaccinia virus in the presence of lipo (L)-IFN-γ95–132, IFN-γ95–132, control peptide, or PBS. Proliferation in the presence of purified inactivated vaccinia virus was determined 48 h later by using Alamar blue dye (25 ). Statistical measurements using the Wilcoxon-Mann-Whitney rank sum test indicated p < 0.05 for mimetic vs control. b, Mice (C57BL/6, n = 5) were immunized using BSA as an Ag in the presence of lipo-IFN-γ95–132, control peptide, or PBS. On the weeks indicated, blood was drawn from mice and measured for the presence of BSA-specific Abs in an ELISA format. The values represent the average with SD. p < 0.05 was obtained for the lipo-mimetic- vs the PBS-treated group.
Measurement of vaccinia-specific cellular response by IFN-γ ELISPOT
CD4 depletion of splenocytes from naive or recovered mice was conducted by using the L3T4 Ab bound to Dynabeads (Invitrogen Life Technologies). ELISPOT assay was conducted by using a kit from Mabtech. Briefly, CD4-depleted cells (105/well) were seeded in a microtiter plate, previously coated with an Ab to IFN-γ, and incubated in the absence or presence of increasing amount of purified vaccinia virus for 24 h at 37°C. After washing, diluted mAb was added and incubated for 2 h, followed by washing and addition of streptavidin-HRP. After 1 h at room temperature, the wells were washed, and TMB substrate was added and optical density read in a plate reader.
FITC labeling and detection of cell and tissue distribution
FITC was conjugated with lipo-IFN-γ95–132, according to the manufacturer’s instruction (Pierce). WISH cells were treated with 5 μM FITC-labeled mimetic or FITC alone for 2 h, followed by visualization in a fluorescent microscope. Mice were injected i.p. with 15 μg of FITC-labeled mimetic or FITC alone. Two hours later, spleen, kidney, lung, and liver were harvested, fixed in formalin and embedded in paraffin. Sections of 7 μm were sliced, mounted on a slide, and viewed in a fluorescent microscope.
Immunofluorescence analysis for determination of nuclear translocation of STAT1α and IFNGR1
WISH cells (3 × 105) were grown overnight on tissue culture-treated slides (Falcon). Cells were treated with 5 μM lipo-IFN-γ95–132, or lipo-IFN-γ95–125 for 6 h. This was followed by permeabilization of cells in 10 mM Tris-HCl (pH 8), TBST for 10 min. Slides were washed in TBS, and nonspecific sites were blocked with 5% nonfat milk in TBS (blocking buffer). Slides were then incubated for 1 h in blocking buffer containing rabbit polyclonal antisera against IFNGR1 (Santa Cruz Biotechnology) and goat polyclonal antisera to human STAT1α (R&D Systems). Cells were washed four times in TBST. This was followed by incubation with secondary Abs, Cy-2-conjugated donkey anti-rabbit (Jackson Immunoresearch Laboratories), and Alexa-Fluor 594-conjugated donkey anti-goat antisera (Molecular Probes) for 1 h. After four washings in TBST, slides were mounted in Prolong antifade solution (Molecular Probes), covered with a coverslip, and sealed with nail varnish. Images were recorded on a Bio-Rad microscope.
Western blot analysis
Cells were washed in PBS and harvested in lysis buffer (50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 0.1% Nonidet P-40, 50 mM NaF, 5 mM EDTA and protease inhibitor mixture; Roche Biochemicals). Protein concentration was measured using a bicinchoninic acid kit from Pierce. Protein (10 μg each) was electrophoresed on an acrylamide gel, transferred to an Immobilon-P filter, and probed with the Abs indicated. HRP-conjugated Abs were used. Detection was by chemiluminescence (Pierce).
Statistical analysis
All experimental data on mice studies were measured for statistical significance by the Kaplan-Meier survival curve and the log rank test with GraphPad Prism software from GraphPad Software.
Results
IFN-mimetic peptide protects mice against lethal doses of vaccinia virus
Vaccinia virus is the prototype of the poxvirus family, and it has been used extensively in mice in the study of the pathogenesis of this family of viruses (15). The IFN-mimetic mIFN-γ95–132 was therefore examined for its ability to protect mice challenged with a lethal dose of the Western Reserve strain of vaccinia virus, which expresses the intact B8R protein (9). Mice (C57BL/6) were treated with 5, 15, 50, or 200 μg of lipophilic (lipo)-mIFN-γ95–132 on days −2, −1, and 0 by i.p. injection. On day 0, the mice were challenged i.p. with 107 PFU of vaccinia virus (Fig. 1 a). This dose is much higher, as compared with the 104 PFU for intranasal administration used by others (16, 17). Complete protection was observed with 200 μg of mimetic peptide, whereas 50, 15, and 5 μg of peptide resulted in 80, 80, and 40% protection, respectively, against death. The recovered mice were healthy over 100 days of the experiment. We have shown that the IFN-mimetic nuclear localization sequence (NLS) located in its C terminus (Materials and Methods) along with an α helix in the N terminus of the peptide are required for mimetic function (18). Further, we showed that cell penetration via the lipophilic palmitate group was required for the function in fibroblasts (11). Thus, control lipophilic peptide lipo-mIFN-γ (95–125), where the NLS is absent, did not protect the mice at 50 or 200 μg, where 100% death occurred by day 8. Also, absent the lipophilic group for cell penetration, 50 or 200 μg of mIFN-γ95–132 did not protect mice against lethal vaccinia virus infection, similar to the PBS control. Thus, lipo-IFN-γ95–132 is highly protective against an overwhelmingly lethal dose of vaccinia virus in mice with requirement of the NLS and cell penetration moieties.
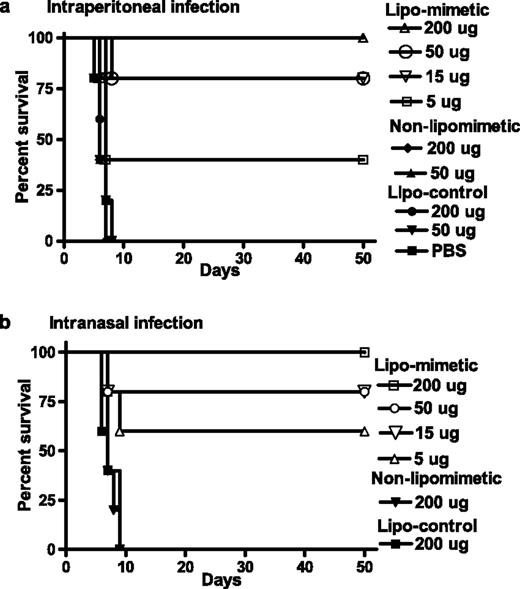
IFN-γ95–132 protects mice against vaccinia virus infection. a, i.p. route of infection with vaccinia virus. C57BL/6 mice (n = 5) were injected i.p. on days −2, −1, and 0 with the following peptides: Lipo-IFN-γ95–132 was given at 200 μg (▵), 50 μg (○), 15 μg (▿), or 5 μg (□). ▪, PBS-injected control. Control peptide lipo-IFN-γ (95–125) was given at 200 μg (•) or 50 μg (▾). Non-lipo-IFN-γ95–132 was given at 200 μg (♦) or 50 μg (▴). On day 0, mice were injected i.p. with 107 PFU of vaccinia virus in a volume of 100 μl with PBS. Survival was followed for >50 days. The significance of difference between different treatment groups was measured by log rank survival method, which gave p values of 0.002, 0.004, 0.009, and 0.03 for administration of 200, 50, 15, and 5 μg per mouse of lipo-mimetic vs the control lipopeptide. b, Intranasal route of infection with vaccinia virus. Mice (n = 5) were injected i.p. with the following peptides. Lipo-IFN-γ95–132 was given at 200 μg (□), 50 μg (○), 15 μg (▿), or 5 μg (▵). Control peptide, lipo-IFN-γ (95–125) (▪), or nonlipophilic IFN-γ95–132 (▾) were given at 200 μg. On day 0, mice were given 106 PFU of vaccinia virus in a volume of 10 μl intranasally. Survival was followed for 50 days. The significance of difference between different treatment groups was measured by log rank survival method, which gave p values of 0.0018, 0.0045, 0.012, and 0.03 for administration of 200, 50, 15, and 5 μg/mouse of lipomimetic vs the control lipo-peptide.
IFN-γ95–132 protects mice against vaccinia virus infection. a, i.p. route of infection with vaccinia virus. C57BL/6 mice (n = 5) were injected i.p. on days −2, −1, and 0 with the following peptides: Lipo-IFN-γ95–132 was given at 200 μg (▵), 50 μg (○), 15 μg (▿), or 5 μg (□). ▪, PBS-injected control. Control peptide lipo-IFN-γ (95–125) was given at 200 μg (•) or 50 μg (▾). Non-lipo-IFN-γ95–132 was given at 200 μg (♦) or 50 μg (▴). On day 0, mice were injected i.p. with 107 PFU of vaccinia virus in a volume of 100 μl with PBS. Survival was followed for >50 days. The significance of difference between different treatment groups was measured by log rank survival method, which gave p values of 0.002, 0.004, 0.009, and 0.03 for administration of 200, 50, 15, and 5 μg per mouse of lipo-mimetic vs the control lipopeptide. b, Intranasal route of infection with vaccinia virus. Mice (n = 5) were injected i.p. with the following peptides. Lipo-IFN-γ95–132 was given at 200 μg (□), 50 μg (○), 15 μg (▿), or 5 μg (▵). Control peptide, lipo-IFN-γ (95–125) (▪), or nonlipophilic IFN-γ95–132 (▾) were given at 200 μg. On day 0, mice were given 106 PFU of vaccinia virus in a volume of 10 μl intranasally. Survival was followed for 50 days. The significance of difference between different treatment groups was measured by log rank survival method, which gave p values of 0.0018, 0.0045, 0.012, and 0.03 for administration of 200, 50, 15, and 5 μg/mouse of lipomimetic vs the control lipo-peptide.
We next conducted the experiment using intranasal challenge of vaccinia virus, the most likely route of transmission of this virus. The challenge dose of vaccinia virus was 106 PFU, ∼100-fold higher than the lethal dose observed by others (16, 17). Similar to i.p. challenge, lipo-IFN-γ95–132 at 200 μg completely protected mice against lethal intranasal vaccinia virus challenge (Fig. 1 b). Eighty percent protection was observed with 50 and 15 μg of mimetic, whereas 60% protection was observed with 5 μg of mimetic. Control peptide, lipo-IFN-γ95–125, and nonlipophilic IFN-γ95–132 did not protect at a 200-μg dose. Thus, whether vaccinia virus was administered i.p. or intranasally, lipo-IFN-γ95–132 was able to completely protect against highly lethal doses of virus.
IFN-mimetic peptide rescues mice from ongoing lethal infection of vaccinia virus
We were interested in determining the ability of lipo-IFN-γ95–132 mimetic to rescue mice from ongoing infection with a lethal dose of vaccinia virus. Accordingly, mice were infected intranasally with 106 PFU of vaccinia virus where 100% death occurs between 6 and 9 days in the absence of the IFN-mimetic peptide. In the mimetic-treated mice, lipo-IFN-γ95–132-mimetic peptide was injected i.p. at 200 μg of peptide for 3 consecutive days beginning at day 1, 2, 3, 4, 5, or 6 after challenge with vaccinia virus. Mice given mimetic up to 2 days after virus challenge were completely protected from the lethal effects of vaccinia virus (Fig. 2), whereas mice treated with mimetic beginning at days 3, 4, and 5 showed 80% protection. Even at day 6 of beginning of treatment post-vaccinia virus infection, when the mice looked very ill with ruffled fur and lethargic appearance, 40% of the mice survived. The recovered mice appeared healthy for the 100 days of observation. Mice were also treated with 200 U of rat IFN-γ at −2, −1, and 0 days relative to virus challenge. Rat IFN-γ was used, because it is active in the mouse and is recognized by the vaccinia B8R protein, whereas mouse IFN-γ interacts with B8R protein at a lower affinity (19, 20). Infected mice were also treated with 200 μg control peptide, lipo-IFN-γ95–125 at −2, −1, and 0 days relative to virus challenge. All of the control peptide and rat IFN-γ-treated mice died by day 9 postinfection. Thus, IFN mimetic can protect mice completely against a lethal dose of vaccinia virus even when treatment was initiated 2 days after infection. Protection was significant, 80% up to 5 days, and 40% even at day 6 of initiation of treatment. IFN mimetic is thus effective against ongoing vaccinia virus infection of ∼100-fold lethal dose.

IFN-γ95–132 protects mice against ongoing vaccinia virus infection. Mice (n = 5) were infected with vaccinia virus (106 PFU in 10 μl) intranasally on day 0. Starting from days 1 (○), 2 (⋄), 3 (▿), 4 (□), 5 (▵), or 6 (•), mice were given 200 μg of lipo-IFN-γ95–132 i.p. in a 100-μl volume with PBS on 3 consecutive days. Control lipo-IFN-γ95–125 (▪) or rat IFN-γ (▾) at (200 U in 100 μl) were administered i.p. on days −2, −1, and 0. Survival was followed for >50 days. p values for the significance of difference between mimetic treatment on days 1, 2, 3, 4, 5, and 6 vs the control peptide treatment were 0.002, 0.002, 0.002, 0.002, 0.012, and 0.02, respectively.
IFN-γ95–132 protects mice against ongoing vaccinia virus infection. Mice (n = 5) were infected with vaccinia virus (106 PFU in 10 μl) intranasally on day 0. Starting from days 1 (○), 2 (⋄), 3 (▿), 4 (□), 5 (▵), or 6 (•), mice were given 200 μg of lipo-IFN-γ95–132 i.p. in a 100-μl volume with PBS on 3 consecutive days. Control lipo-IFN-γ95–125 (▪) or rat IFN-γ (▾) at (200 U in 100 μl) were administered i.p. on days −2, −1, and 0. Survival was followed for >50 days. p values for the significance of difference between mimetic treatment on days 1, 2, 3, 4, 5, and 6 vs the control peptide treatment were 0.002, 0.002, 0.002, 0.002, 0.012, and 0.02, respectively.
Orally administered IFN mimetic protects mice against intranasal challenge with vaccinia virus
All of the protection described above involved i.p. administration of the IFN mimetic. The question arises as to whether orally administered mimetic would similarly protect mice against vaccinia virus. Accordingly, we administered 250-1000 μg lipo-IFN-γ95–132 or lipo-control peptide orally to mice (five per group) via feeding needles at days −2, −1, and 0, after which the mice were challenged intranasally with 106 PFU of vaccinia virus on day 0 (Fig. 3). Mice were completely protected from death by 1000 μg of mimetic, whereas 500 and 250 μg of mimetic resulted in 60 and 40% protection, respectively. Control peptide-fed mice all died by day 9. The peptides were administered in PBS; thus, special treatments such as use of liposomes for delivery or raising the pH to protect against acid proteases of the intestine were not required. These results provide proof-of-concept that the IFN mimetic is protective against intranasal vaccinia virus even when it is administered orally.
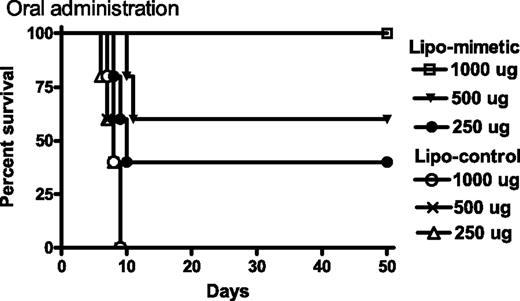
Oral administration of lipo-IFN-γ95–132 protects mice against intranasal challenge with vaccinia virus. Mice (n = 5) on days −2, −1, and 0 were given 1000 μg (□), 500 μg (▾), or 250 μg (•) of lipo-IFN-γ95–132, or 1000 μg (○), 500 μg (×), or 250 μg (▵) of control peptide. On day 0, 106 PFU of vacinia virus were administered intranasally. Survival was followed for >50 days. The significance of difference between different treatment groups was measured by the log rank survival method, which gave p values of 0.002, 0.0027, and 0.04 for administration of 1000, 500, and 250 μg per mouse of lipomimetic vs the control lipopeptide.
Oral administration of lipo-IFN-γ95–132 protects mice against intranasal challenge with vaccinia virus. Mice (n = 5) on days −2, −1, and 0 were given 1000 μg (□), 500 μg (▾), or 250 μg (•) of lipo-IFN-γ95–132, or 1000 μg (○), 500 μg (×), or 250 μg (▵) of control peptide. On day 0, 106 PFU of vacinia virus were administered intranasally. Survival was followed for >50 days. The significance of difference between different treatment groups was measured by the log rank survival method, which gave p values of 0.002, 0.0027, and 0.04 for administration of 1000, 500, and 250 μg per mouse of lipomimetic vs the control lipopeptide.
Comparison of vaccinia virus levels in tissues of mice protected by IFN mimetic vs those treated with control peptide
Related to the protective effect of IFN mimetic, we determined the virus load of selected organs at day 6 of intranasal challenge of mice with 106 PFU of vaccinia virus. As shown in Table I, control peptide-treated mice contained high levels of virus in the respiratory system, 7.5 × 105 PFU in the trachea and 6.5 × 105 in the lungs. Spleen and brain also contained infectious virus, 3.5 × 103 and 4.5 × 103 PFU, respectively. IFN mimetic-treated mice, however, were negative for vaccinia virus in all of these organs at day 6. This suggests that the mimetic is very effective at inhibiting vaccinia virus replication in vivo, which is consistent with the effective protection of mice against the lethal effects of the virus.
Virus levels in tissues of IFN-mimetic and control peptide-treated mice challenged with vaccinia virus
| Tissue | Mimetic-Treated Mice (PFU) | Control-Treated Mice (PFU) |
|---|---|---|
| Trachea | <LOD | 7.5 ± 1.0 × 105 |
| Lung | <LOD | 6.5 ± 0.5 × 105 |
| Spleen | <LOD | 3.5 ± 0.3 × 103 |
| Brain | <LOD | 4.5 ± 0.5 × 103 |
| Tissue | Mimetic-Treated Mice (PFU) | Control-Treated Mice (PFU) |
|---|---|---|
| Trachea | <LOD | 7.5 ± 1.0 × 105 |
| Lung | <LOD | 6.5 ± 0.5 × 105 |
| Spleen | <LOD | 3.5 ± 0.3 × 103 |
| Brain | <LOD | 4.5 ± 0.5 × 103 |
Mice (n = 3) on day 6 after i.p. administration of mimetic or control peptide and intranasal challenge with vaccinia virus were sacrificed, and their organs were used to assay for vaccinia virus. LOD refers to limit of detection, which was 100 PFU/organ.
Potent adaptive immunity develops in mice treated with IFN-mimetic peptide
The question arises as to whether mice protected by the IFN mimetic against lethal virus infection developed adaptive immunity to subsequent infection with a lethal dose of vaccinia virus? We therefore rechallenged mimetic-protected mice 10 wk after the first infection with a second dose of 106 PFU vaccinia administered intranasally without the additional mimetic peptide treatment. All five mice of the rechallenged group were protected against the lethality of vaccinia virus, without showing any symptoms of illness, whereas all five of the naive control mice died by 7–9 days from the vaccinia virus challenge (Fig. 4 a). Thus, the mimetic, in addition to providing protection against the first exposure to vaccinia virus, also allowed the mice to develop protective adaptive immunity to subsequent challenge with the virus.
![FIGURE 4. Adaptive immune response in mice that have recovered from vaccinia virus infection with IFN-γ mimetic treatment. a, Survival of mice (n = 5) against rechallenge with vaccinia virus. Naive mice (▪), or those that had recovered from vaccinia virus infection for 50 days (▾), were infected with vaccinia virus (106 PFU in 10 μl) intranasally. Survival was followed for >50 days. The significance of difference as measured by log rank survival gave p = 0.002 for the rechallenged group vs the naive mice. Cont, Control. b, Cell-mediated immune response in mice recovered from vaccinia virus infection and IFN-γ mimetic treatment. Splenocytes (105) from naive or recovered mice (n = 3), 2 or 3 wks after infection and mimetic treatment, were incubated with UV-inactivated purified vaccinia virus. Four days later, [3H]thymidine was added for 8 h, and its incorporation was followed. Stimulation index refers to the incorporation in splenocytes cultured with test Ag divided by incorporation in splenocytes cultured with medium alone. The averages with SD are shown. c, Vaccinia virus-specific response in CD4-depleted splenocytes by ELISPOT analysis. Splenocytes from naive or recovered mice (n = 3) 2 or 3 wks after infection and IFN mimetic treatment were depleted of CD4 cells by using appropriate Abs, as described in Materials and Methods, and 105 cells thus obtained were incubated in microtiter plates, previously coated with Ab to IFN-γ, with the following PFU of vaccinia virus: 5 × 104 (column 1); 15 × 104 (column 2); and 50 × 104 (column 3). After 2 days of incubation, the number of spots (IFN-γ-secreting cells) per well were counted. The numbers represent the average with SD. d and e, Presence of vaccinia specific Abs in mice recovered from infection and mimetic treatment. Sera collected from mice (n = 5) during 36 days were diluted and added to wells of microtiter plates coated with UV-inactivated vaccinia virus. After washing to remove nonspecific binding, secondary anti-mouse IgM (d) or IgG (e) Ab conjugated to HRP was added, followed by addition of o-phenylenediamine substrate and optical density measurement. Dilutions of serum for IgM were 1/100, 1/200, and 1/500 in columns 1, 2, and 3, respectively, on the days indicated in d. Dilutions of serum for IgG were 1/1,000, 1/10,000, and 1/30,000 in the three columns, respectively, on the days indicated in e. The values represent the average with SD. f, Neutralizing Abs in recovered mice. Sera taken from mice (n = 5) on the days indicated were diluted 1/20, 1/50, and 1/500, respectively, mixed with 100 PFU of vaccinia virus, and incubated for 1 h, followed by addition to BSC-40 cells. Reduction in the number of plaques is shown as percentage with SD.](https://aai.silverchair-cdn.com/aai/content_public/journal/jimmunol/178/7/10.4049_jimmunol.178.7.4576/2/m_zim0070749360004.jpeg?Expires=1716291636&Signature=2iLHtvIiaWEIXdIn-H8uFl4m6cfvkwPWwC2sZNE4kB~O8ZJ4AuY8yctenh3-KUEOYhzH7iKHOI4l0Ug7BNUuKaLhP5UDmgf0pvWD~MDOlQUO93x3kskn8iTNgKIHklBZLx1yM~nn6KyZEP1q5ZAjV1kwOhrRCfouTBJDYsrqssjBHVQo317MEtCPefKugT7AywiN-Nxh-BFfjRac52Nbao~MIStZOBOWSK7Qst17SO9aBJrkc7pVJ4BJEsXWzk0eQoscKpYDkeq6gESbnBw1R4DNMPNmvSb7762tY3cV7vMqt4591OvroDA1iXX5mEiPiiSksd5njaZK4gRCKl2GrA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Adaptive immune response in mice that have recovered from vaccinia virus infection with IFN-γ mimetic treatment. a, Survival of mice (n = 5) against rechallenge with vaccinia virus. Naive mice (▪), or those that had recovered from vaccinia virus infection for 50 days (▾), were infected with vaccinia virus (106 PFU in 10 μl) intranasally. Survival was followed for >50 days. The significance of difference as measured by log rank survival gave p = 0.002 for the rechallenged group vs the naive mice. Cont, Control. b, Cell-mediated immune response in mice recovered from vaccinia virus infection and IFN-γ mimetic treatment. Splenocytes (105) from naive or recovered mice (n = 3), 2 or 3 wks after infection and mimetic treatment, were incubated with UV-inactivated purified vaccinia virus. Four days later, [3H]thymidine was added for 8 h, and its incorporation was followed. Stimulation index refers to the incorporation in splenocytes cultured with test Ag divided by incorporation in splenocytes cultured with medium alone. The averages with SD are shown. c, Vaccinia virus-specific response in CD4-depleted splenocytes by ELISPOT analysis. Splenocytes from naive or recovered mice (n = 3) 2 or 3 wks after infection and IFN mimetic treatment were depleted of CD4 cells by using appropriate Abs, as described in Materials and Methods, and 105 cells thus obtained were incubated in microtiter plates, previously coated with Ab to IFN-γ, with the following PFU of vaccinia virus: 5 × 104 (column 1); 15 × 104 (column 2); and 50 × 104 (column 3). After 2 days of incubation, the number of spots (IFN-γ-secreting cells) per well were counted. The numbers represent the average with SD. d and e, Presence of vaccinia specific Abs in mice recovered from infection and mimetic treatment. Sera collected from mice (n = 5) during 36 days were diluted and added to wells of microtiter plates coated with UV-inactivated vaccinia virus. After washing to remove nonspecific binding, secondary anti-mouse IgM (d) or IgG (e) Ab conjugated to HRP was added, followed by addition of o-phenylenediamine substrate and optical density measurement. Dilutions of serum for IgM were 1/100, 1/200, and 1/500 in columns 1, 2, and 3, respectively, on the days indicated in d. Dilutions of serum for IgG were 1/1,000, 1/10,000, and 1/30,000 in the three columns, respectively, on the days indicated in e. The values represent the average with SD. f, Neutralizing Abs in recovered mice. Sera taken from mice (n = 5) on the days indicated were diluted 1/20, 1/50, and 1/500, respectively, mixed with 100 PFU of vaccinia virus, and incubated for 1 h, followed by addition to BSC-40 cells. Reduction in the number of plaques is shown as percentage with SD.
Adaptive immune response in mice that have recovered from vaccinia virus infection with IFN-γ mimetic treatment. a, Survival of mice (n = 5) against rechallenge with vaccinia virus. Naive mice (▪), or those that had recovered from vaccinia virus infection for 50 days (▾), were infected with vaccinia virus (106 PFU in 10 μl) intranasally. Survival was followed for >50 days. The significance of difference as measured by log rank survival gave p = 0.002 for the rechallenged group vs the naive mice. Cont, Control. b, Cell-mediated immune response in mice recovered from vaccinia virus infection and IFN-γ mimetic treatment. Splenocytes (105) from naive or recovered mice (n = 3), 2 or 3 wks after infection and mimetic treatment, were incubated with UV-inactivated purified vaccinia virus. Four days later, [3H]thymidine was added for 8 h, and its incorporation was followed. Stimulation index refers to the incorporation in splenocytes cultured with test Ag divided by incorporation in splenocytes cultured with medium alone. The averages with SD are shown. c, Vaccinia virus-specific response in CD4-depleted splenocytes by ELISPOT analysis. Splenocytes from naive or recovered mice (n = 3) 2 or 3 wks after infection and IFN mimetic treatment were depleted of CD4 cells by using appropriate Abs, as described in Materials and Methods, and 105 cells thus obtained were incubated in microtiter plates, previously coated with Ab to IFN-γ, with the following PFU of vaccinia virus: 5 × 104 (column 1); 15 × 104 (column 2); and 50 × 104 (column 3). After 2 days of incubation, the number of spots (IFN-γ-secreting cells) per well were counted. The numbers represent the average with SD. d and e, Presence of vaccinia specific Abs in mice recovered from infection and mimetic treatment. Sera collected from mice (n = 5) during 36 days were diluted and added to wells of microtiter plates coated with UV-inactivated vaccinia virus. After washing to remove nonspecific binding, secondary anti-mouse IgM (d) or IgG (e) Ab conjugated to HRP was added, followed by addition of o-phenylenediamine substrate and optical density measurement. Dilutions of serum for IgM were 1/100, 1/200, and 1/500 in columns 1, 2, and 3, respectively, on the days indicated in d. Dilutions of serum for IgG were 1/1,000, 1/10,000, and 1/30,000 in the three columns, respectively, on the days indicated in e. The values represent the average with SD. f, Neutralizing Abs in recovered mice. Sera taken from mice (n = 5) on the days indicated were diluted 1/20, 1/50, and 1/500, respectively, mixed with 100 PFU of vaccinia virus, and incubated for 1 h, followed by addition to BSC-40 cells. Reduction in the number of plaques is shown as percentage with SD.
Various immunological parameters were examined in the combined vaccinia virus and IFN mimetic-treated mice. Splenocytes obtained 2 or 3 wk after virus challenge from protected mice as well as from naive mice were incubated with purified UV-inactivated vaccinia virus in a proliferation assay. The protected mice had stimulation indices of 4.5 and 8.5, respectively, at 2 and 3 wk postchallenge with the virus (Fig. 4,b). Splenocytes from the naive mice did not respond to the virus. The proliferation results are suggestive of induction of vaccinia virus-specific CD4 T cells. Splenocytes were also tested for the production of the cytokine IFN-γ in an ELISPOT assay. Similar to the proliferation response, CD4-depleted splenocytes (105/well) exposed to vaccinia virus showed increased secretion of IFN-γ by ELISPOT at 2 and 3 wk after virus challenge (Fig. 4 c), which suggests the induction and/or activation of cytotoxic CD8 T cells to vaccinia virus. Naive cells by comparison produced negligible amounts of IFN-γ when stimulated with virus, which again is evidence that the initial aborted infection induced an adaptive immune response.
Sera from protected mice were examined over 36 days for Abs to vaccinia virus in an ELISA with purified virus. As shown in Fig. 4,d, the IgM Ab response peaked at day 11, which was complemented by the later peaking of the IgG Ab response (Fig. 4,e), indicating that the humoral arm of the immune system was also induced in the IFN mimetic peptide-protected vaccinia virus-infected mice. The Ab response also resulted in the production of neutralizing Abs that peaked at days 11 through 36 and probably involved both IgM and IgG Abs to vaccinia virus as shown in Fig. 4 f. Thus, the mimetic-protected mice mounted both a cellular and humoral immune response to vaccinia virus. Apparently, the virus challenge or the early phases of virus replication provided sufficient viral Ag load for the vigorous protective immune response under conditions where the IFN-mimetic peptide provided protection against the lethality of the virus.
IFN-mimetic IFN-γ95–132 possesses adjuvant activity
We were interested in determining whether the IFN mimetic possessed adjuvant activity that may have contributed to the anti-vaccinia immune response in protecting mice. Thus, mice were infected with a sublethal dose of vaccinia virus in the presence of mimetic or control peptide, and the cellular and humoral immune response to virus were monitored. Mice were injected i.p. with peptides at days −2, −1, and 0 relative to the intranasal challenge with 104 PFU of vaccinia virus. Proliferation in the presence of vaccinia virus was determined with splenocytes at 4 wk postinfection. As shown in Fig. 5 a, both lipo-IFN-γ95–132- and nonlipo-IFN-γ95–132-treated murine splenocytes showed greater proliferation in the presence of vaccinia virus than those of the control peptide-treated mice, with the lipophilic form being more effective. An unrelated peptide from residues 253 to 287 from IFNGR1 with a lipophilic residue was used as a control peptide in this experiment. Thus, the mimetic in either a lipophilic state for cell penetration or in a nonlipophilic state enhanced the proliferation of splenocytes from mice infected with vaccinia virus.
Vaccinia virus is a potent Ag and thus may possess intrinsic adjuvant activity. This in turn could mask some of the adjuvancy of the mimetic. We thus determined the adjuvancy of the IFN mimetic against a soluble protein, BSA, focusing on the humoral response. As shown in Fig. 5 b, lipomimetic-treated mice showed significant increases in anti-BSA IgG Ab by ELISA at weeks 2 through 4 when compared with the BSA treatment alone. Mice treated with lipo-control peptide from residues 95 to 106 had Ab responses similar to that of BSA alone. The IFN mimetic thus possesses adjuvant effects against vaccinia virus and BSA Ags.
Tissue distribution of lipo-IFN-γ95–132 peptide in mice
To test the ability of lipo-IFN-γ95–132 to penetrate into cells in culture and to study its biodistribution in various tissues of mice, FITC-conjugated lipo-IFN-γ95–132 was prepared. After 2 h of treatment of WISH cells with 5 μM FITC-lipo-IFN-γ95–132, visualization in a fluorescent microscope revealed that this peptide was internalized (Fig. 6), whereas FITC alone did not show any accumulation in the cells. This is consistent with the cell-penetrating palmitate lipophilic group complexed to IFN-γ95–132. FITC-lipo-IFN-γ95–132 concentrated in the nucleus, consistent with the functional NLS in the C terminus of the mimetic (21). Next, we monitored tissue distribution of FITC-conjugated mimetic in mice injected with 15 μg of FITC-lipo-IFN-γ95–132. An equivalent amount of unconjugated FITC was also injected in different mice. Tissue distribution was ascertained 2 h later for FITC-mimetic and FITC alone from sections of paraffin-embedded tissues and cells by fluorescent microscopy (Fig. 6). FITC-lipo-IFN-γ95–132 was internalized in peritoneal cells of injected mice and was present above FITC background in spleen, liver, lung, and kidney. Thus, i.p. injected IFN mimetic is disseminated to various tissues where it may help counteract vaccinia virus infection.

Tissue distribution of IFN-γ95–132 peptide. A 5-μM concentration of FITC-conjugated lipo-IFN-γ95–132 or an equivalent amount of free FITC were added to WISH cells for 2 h, followed by visualization in a fluorescent microscope. Similarly, 15 μg of FITC-lipo-mimetic or an equivalent amount of free FITC were injected i.p. into different mice. Two hours later, peritoneal cells and the organs indicated were harvested, embedded in paraffin, sectioned, and viewed in a fluorescent microscope.
Tissue distribution of IFN-γ95–132 peptide. A 5-μM concentration of FITC-conjugated lipo-IFN-γ95–132 or an equivalent amount of free FITC were added to WISH cells for 2 h, followed by visualization in a fluorescent microscope. Similarly, 15 μg of FITC-lipo-mimetic or an equivalent amount of free FITC were injected i.p. into different mice. Two hours later, peritoneal cells and the organs indicated were harvested, embedded in paraffin, sectioned, and viewed in a fluorescent microscope.
IFN mimetic induces nuclear translocation of IFNGR1 and activation of STAT1α
To test the mechanism of action of IFN mimetic, WISH cells treated with IFN mimetic or a control peptide were doubly immunostained with Abs to STAT1α and one of the receptor subunits of IFN-γ receptor, IFNGR1. Treatment of these cells with IFN mimetic resulted in the simultaneous nuclear translocation of the transcription factor STAT1α and the receptor subunit, IFNGR1 (Fig. 7,a), as was observed previously by us with secreted or nonsecreted IFN-γ (21, 22). The control peptide showed diffused appearance of these molecules in the cell. Treatment with IFN mimetic also resulted in phosphorylation of STAT1α, which was not observed in the untreated or control peptide treated cells (Fig. 7 b) under conditions where equivalent amounts of the total STAT1α protein were analyzed on the gel. Therefore, IFN-γ mimetic acts through a mechanism similar to that of intact IFN-γ in activation of STAT1α. Related to this, nuclear IFNGR1 was recently shown to bind to the GAS element at the promoter site of IFN-γ-activated genes where it functioned as a trans activator (22).
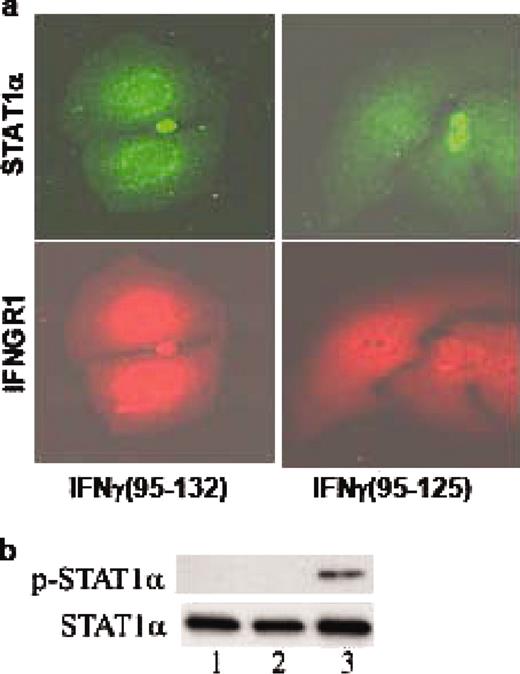
IFN-γ mimetic treatment results in activation of STAT1α and nuclear translocation of STAT1α and IFNGR1. a, Nuclear translocation of STAT1α and IFNGR1. WISH cells treated with 10 μM lipo-IFN-γ95–132 (left columns), or lipo-IFN-γ (95–125) (right columns) were stained simultaneously with Abs to STAT1α and IFNGR1. Secondary Abs to STAT1α conjugated to Alexa 594 (top row), or to IFNGR1 conjugated to Cy-2 (bottom row) were used and analyzed by fluorescence microscopy. b, Phosphorylation of STAT1α by IFN mimetic. Cell extracts from WISH cells, untreated (lane 1), control peptide treated (lane 2), or IFN mimetic treated (lane 3) were electrophoresed, transferred to Immobilon-P, and probed with an Ab to phospho-STAT1α (top row). The filter was stripped and reprobed with an Ab for total STAT1α (bottom row) to ensure equal loading of protein.
IFN-γ mimetic treatment results in activation of STAT1α and nuclear translocation of STAT1α and IFNGR1. a, Nuclear translocation of STAT1α and IFNGR1. WISH cells treated with 10 μM lipo-IFN-γ95–132 (left columns), or lipo-IFN-γ (95–125) (right columns) were stained simultaneously with Abs to STAT1α and IFNGR1. Secondary Abs to STAT1α conjugated to Alexa 594 (top row), or to IFNGR1 conjugated to Cy-2 (bottom row) were used and analyzed by fluorescence microscopy. b, Phosphorylation of STAT1α by IFN mimetic. Cell extracts from WISH cells, untreated (lane 1), control peptide treated (lane 2), or IFN mimetic treated (lane 3) were electrophoresed, transferred to Immobilon-P, and probed with an Ab to phospho-STAT1α (top row). The filter was stripped and reprobed with an Ab for total STAT1α (bottom row) to ensure equal loading of protein.
Discussion
Poxviruses are highly adept at evading the host innate immune response due to their many poxvirus evasion genes and their resultant protein products (15). There are, for example, >18 proteins produced by poxviruses that interfere with different aspects of the host defense, including the actions of IFNs, TNF, various chemokines, and interleukins among others. Poxvirus proteins that interfere with IFN function are of particular interest in assessing strategies to circumvent or overcome poxvirus infections. Vaccinia virus, for example, produces decoy receptors to major IFNs, such as IFNα and IFNβ (types I) and IFN-γ (type II) that bind to the IFNs and inhibit their interaction with the extracellular domain of their respective receptors (15). The B8R gene of vaccinia virus encodes a 43 kDa glycoprotein that is secreted as a homodimer from infected cells early in infection (9). This protein is a homolog of the extracellular domain of the IFN-γ receptor chain IFNGR1 and thus inhibits IFN-γ function by binding to it and blocking its access to the IFN-γ receptor on cells. Thus, the challenge is to develop IFN-like antivirals that overcome poxvirus virulence factors such as B8R protein.
We have developed small peptide mimetics of IFN-γ, based not on the classical model of IFN-γ-initiated signaling by extracellular interaction but rather on direct intracellular signaling by IFN-γ. IFN-γ, its receptor subunit IFNGR1, and transcription factor STAT1α are transported to the nucleus of cells as a complex where IFN-γ provides a classical polycationic NLS for such transport (23). The C terminus of IFN-γ, represented here by the mouse IFN-γ peptide, IFN-γ95–132, was capable of also forming a complex with IFNGR1 and STAT1α when introduced intracellularly and provided the NLS signaling for nuclear transport (24). Importantly, mouse IFN-γ95–132 and human IFN-γ95–134 mimetics both induced an antiviral state and up-regulation of MHC class I molecules in cells similar to that of full length IFN-γ (12). Both IFN-γ and its peptide mimetics bind to an intracellular site, IFNGR1253–287, on the cytoplasmic domain of receptor subunit IFNGR1. This binding plays a role in tyrosine phosphorylation events, catalyzed by JAK1 and JAK2 kinases that result in the phosphorylation and binding of STAT1α to the cytoplasmic domain of IFNGR1. Important structural requirements for IFN-γ-mimetic activity are a polycationic NLS and an α helix in the mimetics (10, 18, 25). Chromatin immunoprecipitations and reporter gene studies of IFN-γ and IFN-γ mimetic-treated cells indicate that they, along with IFNGR1 and STAT1α, bind to the IFN-γ activation site element of IFN-γ-activated genes and participate in STAT1α-mediated transcription (22). IFN-γ intracellular events played the key role in development of IFN-γ mimetics (6, 26). In contrast to intact IFN-γ, therefore, the mimetics do not bind to poxvirus B8R protein and can thus initiate an antiviral response in the presence of B8R protein in cell culture models of vaccinia virus infections (12), as well as mouse models of encephalomyocarditis virus infections (27).
In the studies presented here, we showed that IFN-γ95–132, containing the lipophilic group palmitate for membrane penetration, protected C57BL/6 mice against highly lethal doses of vaccinia virus under a variety of conditions of infection. The i.p. injection of mimetic before and at the time of intranasal (106 PFU) or i.p. (107 PFU) challenge with virus resulted in complete protection of mice at 200 μg of mimetic, 80% with 15 and 50 μg, and 40 to 60% protection with 5 μg of IFN-mimetic peptide. Importantly, initiation of treatment of mice with mimetic, 200 μg/day, at day 2 postinfection with 106 PFU (intranasally) of vaccinia virus resulted in no fatalities compared with 100% for controls, and initiation of treatment as late as day 6 resulted in recovery of 40% of the infected mice. Vaccinia virus B8R protein has been reported to bind to mouse IFN-γ with relatively low affinity when compared with human IFN-γ with dissociation constants, Kd, of 4 × 10−8 M vs 9 × 10−11 M (19, 20). The fact that vaccinia virus vectors and vaccinia with inactivated B8R protein are attenuated in their virulence for mice, would suggest that the low affinity interaction is still biologically significant (20, 28). This assumes the production of relatively high concentrations of B8R protein in vivo by vaccinia virus, which would enhance the binding to and neutralization of mouse IFN-γ involving the law of mass action (29). Thus, by bypassing the virulence factor B8R, the IFN-γ mimetic was able to convert an aggressive infection in mice into a rather benign infection.
In addition to the direct antiviral properties, IFN-γ95–132 also had an adjuvant effect on both the humoral and cellular immune response. Mimetic enhanced the IgG Ab response to both vaccinia virus and BSA. Cellular immunity to vaccinia virus was also enhanced as determined by splenocyte proliferation. The adjuvancy of the mimetic was observed with or without the lipophilic group attached. Thus, it does not depend on mimetic penetration of the plasma membrane of nonphagocytic cells. However, given that uptake of the mimetic by APCs does not depend on the lipophilic group, internalization by these cells could play an important role in the enhanced immune response.
It has recently been reported that insertion of Th2 cytokine IL-4 gene into several different poxviruses resulted in significantly enhanced pathogenesis in animal models of poxvirus infection (30). Specifically, ectromelia virus containing the IL-4 gene converted genetically resistant and previously immunized C57BL/6 mice to complete susceptibility and increased the pathogenesis of ectromelia virus in the already susceptible BALB/c mouse strain (30). Interestingly, cidofovir did not protect C57BL/6 mice infected with ectromelia-IL-4 virus against death (31). Similar enhanced pathology was observed in resistant rabbits infected with myxoma virus in which the IL-4 gene was inserted (32). An analysis of cytokines, Th1, Th2, and CTL responses suggested that the IL-4 negatively affected the Th1 cytokine IFN-γ and CTL. As we have shown above, Th2 response as per neutralizing Abs to poxvirus is not necessarily obtained at the expense of CTL responses. The IFN-γ mimetic-treated mice had a strong cellular and neutralizing Ab response. Furthermore, the mimetic had adjuvant effects on both the humoral and cellular immune responses. The protective effects of mimetic here then are in sharp contrast to the enhanced susceptibility in animals infected with the poxvirus-IL-4 construct.
Nucleoside analogs such as cidofovir have been the most promising chemotherapeutics for poxvirus infections (31). Cidofovir, however, has undesirable toxic side effects, most notably involving the kidneys because of a lack of virus-specific effects. More recently, protein tyrosine kinases have been targeted in animal models of vaccinia virus infection. Poxvirus pathogenesis is aided by endogenous EGF-like growth factors with production of smallpox growth factor by variola virus and vaccinia virus growth factor by vaccinia virus (33). Drugs that inhibit the cellular Erb-1 signal transduction pathway, CI-1033 and related 4-anilinoquinazolines, inhibited smallpox growth factor-induced signaling, blocked vaccinia virus release from infected cells, and prolonged the life of mice infected with lethal vaccinia virus infection (16). Similarly, the abl kinase inhibitor, Gleevec, also inhibited vaccinia virus release from cells and extended the life of mice against lethal vaccinia virus infection (17). The amount of vaccinia virus used in the mouse studies with kinase inhibitors was 100-fold less than used in our mimetic studies here. Postinfection therapy was not reported at all for Gleevec (17), and CI-1033 treatment alone of mice starting 2 days after challenge with 2 × 104 PFU of virus was ineffective (16). CI-1033 did augment postinfection therapy with anti-vaccinia Abs (16). Finally, none of the tyrosine kinase inhibitors have been tested for their effect on the immune system, so we do not know whether they have adjuvant effects or suppressive effects on poxvirus immunity. Thus, the data presented in this report demonstrate that the IFN-γ-mimetic peptide is a potent inhibitor of highly lethal doses of vaccinia virus in mice even when administered well into ongoing disease. The mimetic also augments the immune response of infected mice to vaccinia virus and thus possesses adjuvant effects. These results suggest that the IFN mimetic is a potential therapeutic for poxvirus infections.
Acknowledgments
We thank Donna Williams for help with microscopy, Tracy Clarke for sectioning of tissues, and S. M. Haider for peptide synthesis.
Disclosures
The authors have no financial conflict of interest.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by National Institutes of Health Grant AI 56152 and Department of Defense Grant W911NF-05-1-0170 (both to H.M.J.).
Abbreviations used in this paper: mIFN-γ, murine IFN-γ; NLS, nuclear localization sequence.
